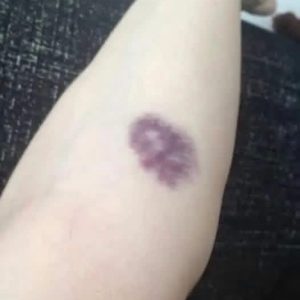
It started as a few harmless-looking red spots on my face, but within hours, the burning pain became absolutely unbearable. My skin felt like it was literally on fire, radiating a heat that no cold compress could extinguish. Doctors were baffled, my family was terrified, and I felt as though my body had suddenly turned against me in a violent, systemic revolt. What I thought was a simple allergic reaction spiraled into a medical mystery that would force me to confront a rare, agonizing, and potentially deadly diagnosis that most people have never even heard of.
The journey to my diagnosis began on an ordinary afternoon when the first signs of the condition—painful, inflamed, and bright red lesions—appeared abruptly on my face and neck. The speed of the onset was alarming. Within hours, these patches of skin grew raised, tender to the touch, and increasingly angry. After a frantic trip to my primary care physician, I was urgently referred to a dermatologist, as the clinical presentation was unlike anything the local clinic had encountered. The immediate suspicion, given my recent history, was a severe drug-related reaction, and my doctors acted quickly to discontinue any medication that might be fueling this aggressive inflammatory response.
The following days were a blur of medical tests and mounting anxiety. My medical team initiated a rigorous workup, which included a skin biopsy, comprehensive blood counts, and specialized testing for auto-antibodies and the lupus anticoagulant to rule out more common systemic diseases. While we waited for the results, my doctors decided to start me on a course of oral corticosteroids. It was a gamble, but it was the right one; within 48 hours, the miracle I had been praying for began to manifest. The agonizing pain that had kept me awake for nights on end started to subside, and the angry, erythematous lesions began to flatten and fade.
When the laboratory results finally returned, they painted a complex picture. I was suffering from leukocytosis, an abnormally high white blood cell count, specifically characterized by neutrophilia. My tests for lupus anticoagulant and certain antibodies were positive, while routine serology remained negative. However, the definitive answer didn’t arrive until the pathology report from the skin biopsy was finalized. The diagnosis was confirmed: Sweet syndrome, or acute febrile neutrophilic dermatosis.
Sweet syndrome is an incredibly rare and often misunderstood inflammatory condition. It is characterized by the sudden appearance of painful, red papules or plaques caused by a dense infiltration of neutrophils—a type of white blood cell—into the skin. The name sounds almost harmless, but the reality of the condition is anything but. The exact mechanism behind why the body suddenly decides to flood the skin with these cells remains a subject of intense scientific study, though current theories suggest it involves a complex, cytokine-driven activation of neutrophils. Essentially, it is an immune-mediated hypersensitivity reaction that can be triggered by a variety of environmental and biological factors, including recent infections, underlying malignancies, or, in many cases, a reaction to specific pharmaceutical agents.
Learning that my condition might be drug-induced was a jarring realization. Doctors explained that Sweet syndrome is frequently linked to a surprising range of common medications, including certain antibiotics, antiepileptics, hormonal contraceptives, antihypertensives, and even some vaccines or colony-stimulating factors. It is a sobering reminder of how our bodies can sometimes overreact to substances meant to heal or support us. The first-line treatment, which I had already begun, involves a high-dose course of oral corticosteroids. While the treatment is often effective at inducing rapid clinical improvement, the psychological impact of surviving such an aggressive, unexplained inflammatory event is profound.
The diagnostic process for Sweet syndrome is notoriously difficult because it mimics so many other common skin disorders. Throughout my treatment, my doctors had to rule out a laundry list of possibilities, including urticaria (hives), contact dermatitis, various forms of toxicoderma, and cutaneous lupus. Because the symptoms are so similar to these more common ailments, a definitive diagnosis requires a perfect, often tedious, correlation between the clinical presentation, exhaustive laboratory work, and the final word from histopathology. This multi-layered diagnostic approach is essential, not just to confirm the presence of Sweet syndrome, but to rule out systemic diseases that might be hiding behind the skin manifestations.
Reflecting on my experience, I realized how much we take the health of our skin for granted. We often think of the skin as merely a protective barrier, yet it is intimately connected to our immune system, our blood chemistry, and our internal organs. When something goes wrong beneath the surface, the skin is often the first to sound the alarm, and in my case, it was screaming for help.
My recovery was a testament to the importance of persistent medical investigation. Had I ignored those initial red spots, hoping they would simply fade away, the inflammation could have continued to escalate, leading to more systemic complications or permanent scarring. The experience served as a powerful lesson in self-advocacy and the critical necessity of seeking specialized dermatological care when a skin issue deviates from the norm. Today, as I continue to monitor my health and manage the lasting awareness of how my immune system functions, I am reminded that our bodies are incredibly resilient. Sweet syndrome may be a rare and frightening visitor, but it is one that, with the right medical guidance, can be understood, treated, and ultimately conquered. The memory of that burning pain remains, but it has been replaced by a deep, unwavering appreciation for the quiet, healthy comfort of my own skin.